Models for proportion (part to whole percent) data
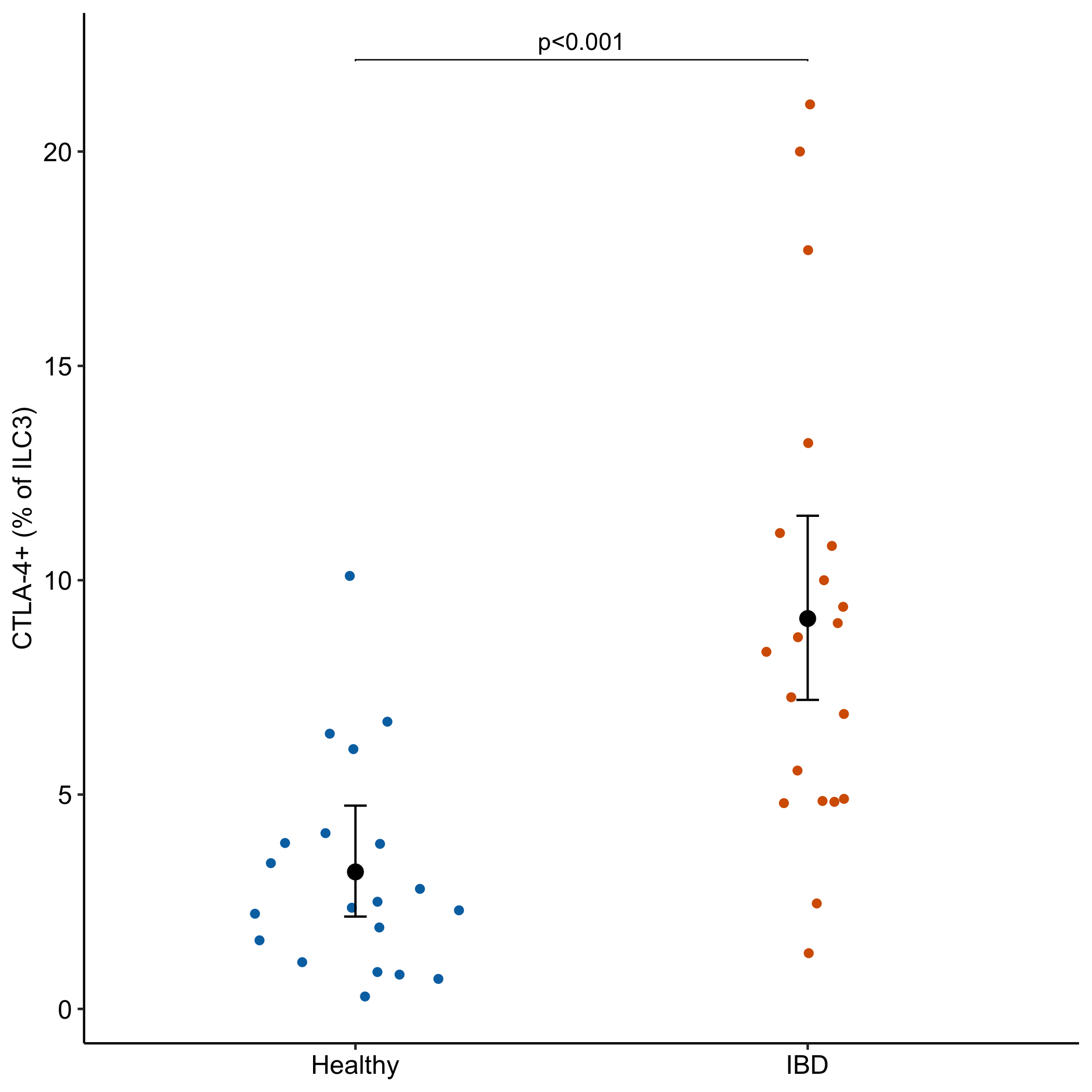
Data From Fig 5c – CTLA-4-expressing ILC3s restrain interleukin-23-mediated inflammation
The data in fig 5c are the percent of a cell subtype relative to all cells of a type, which is a common response variable in bench biology. The researchers analyzed this part-to-whole (a proportion) response with a Mann-Whitney U test. A better practice method is a GLM of the count of the part using the count of the whole as an offset. The effect is the ratio of the treatment proportion relative to the control proportion. This post also shows how to plot the proportion with estimated means and CIs from the GLM with offset model.
percents
proportions
generalized linear model
offset
power
simulation
Author
Jeff Walker
Published
July 5, 2024
Modified
October 2, 2024
Better than Reproducibility of Fig 5c. The CIs and p-values are from a quasipoisson GLM model with offset
Should I use Mann-Whitney, a Linear Model, or a Quasibinomial GLM for proportion data?
Answer: None of the Above
Proportions have strange, non-normal distributions. Samples tend to be right skewed if the mean is closer to zero but left skewed if the mean is closer to 1. And, samples with means closer to 0.5 tend to have more variance than samples closer to 0 or 1. This is probably why the authors used a non-parametric test (Mann-Whitney U). A Mann-Whitney test has higher power than a linear model / t-test but other than the p-value, there are no statistics to plot with the data. There is no effect in a a Mann-Whitney – it is not a test of the differences in the means (or medians!). And, we can’t compute a confidence interval for the group means.
A proportion is a ratio of a part to the whole. A modern statistical method for analyzing proportion data is to use a log-link generalized linear model with the part as the response and the whole as an offset variable. An offset variable is a covariate but the coefficient is forced to be one. By forcing the coefficient to be one, the effect is the ratio of the means of the two proportions (or the difference of the means of the proportions in log space).
GLM for percent (proportion of whole) data when both part and whole are known
The effect (2.85) is the ratio of the mean proportion of the IBD group to the mean proportion of the Health group.
Code
# pretending to have the full count data. Don't do this if you don't have it!fake_ilc3 <-432987fig5c[, ctla4_count := ctla4_pos *432987]fig5c[, ilc3_count :=432987]m3 <-glm(ctla4_count ~ health +offset(log(ilc3_count)),family =quasipoisson(link ="log"),data = fig5c)m3_emm <-emmeans(m3, specs ="health", type ="response")m3_pairs <-contrast(m3_emm, method ="revpairwise") |>summary(infer =TRUE)m3_pairs |>kable() |>kable_styling(full_width =FALSE)
For data that look like those in fig. 5c, the Mann-Whitney has good Type I control and high power relative to the linear model/t-test and compared to the quasibinomial GLM model. But the quasipoisson GLM offset model for the original counts has 20% higher power than the Mann-Whitney, but slightly elevated Type 1. And the beauty of the quasipoisson GLM is we get model statistics to plot with the data (see the plot below).
The table below shows the Power and Type I error when alpha is set to 0.03. The power for the quasipoisson model with alpha = 0.03 has good Type I control and is 13% higher than the power for the Mann-Whitney with alpha = 0.05.
If using the quasipoisson GLM we could make the test a bit more conservative by using smaller alpha, but again, I don’t think bench biologists really use alpha in any formal sense.
Plot the model
A good question is, how do we Plot the Model? A proportion response makes sense for communication but the emmeans table has whole-count adjusted counts as the mean response. But these adjusted counts are proportions, just unscaled by the denominator. So we can rescale the mean and CIs as proportions (or percents) by dividing by the mean counts of the whole (ilc3 count).